Spontaneous penetration of carbon nanotubes through lipid bilayers: A computational perspective
Abstract
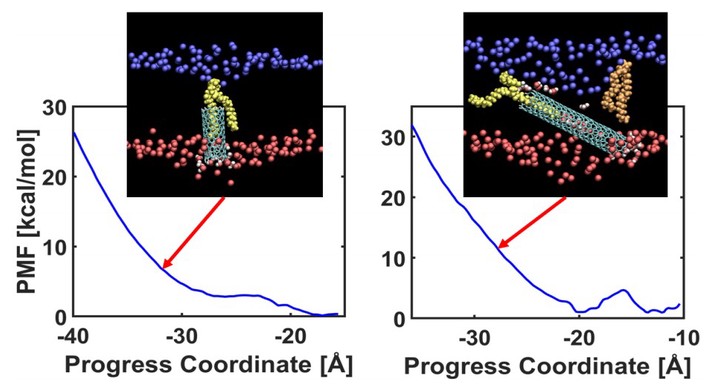
Carbon nanotubes (CNTs) have been widely explored for applications ranging from drug delivery to energy conversion. To evaluate their potential as drug-delivery agents, it is essential to understand the molecular mechanisms governing their interactions with, and possible transport across, cell membranes. Molecular dynamics (MD) simulations provide a powerful framework for probing these processes at molecular resolution. In this study, we investigate the spontaneous membrane insertion and deep penetration of pristine CNTs (p-CNTs) using the weighted ensemble (WE) method combined with all-atom MD simulations. Two p-CNTs with similar diameters but different lengths were examined to elucidate how nanotube length influences membrane-interaction behavior. Both p-CNTs readily insert into the hydrophobic core of the membrane; however, they exhibit distinct behaviors near the lower leaflet. Although neither p-CNT fully escapes into the opposite aqueous phase due to strong hydrophobic interactions, the WE approach enables detailed characterization of their penetration pathways and the associated free-energy landscape within the bilayer.