Electrolytes for Li-ion Transport and Materials Design
 Molecular dynamics (MD) simulation of LiTFSI + water
Molecular dynamics (MD) simulation of LiTFSI + water
We are studying the characteristics of electrolytes used in lithium-ion batteries to design improved materials based on these properties. To achieve this, we employ density functional theory calculations and molecular dynamics simulations to explore how lithium ions move within different electrolytes.
In addition, our research includes investigating the functional and mechanical properties of various materials. Through modeling and simulations, we aim to gain a comprehensive understanding of these properties.
Publications
Ab Initio Insights into Diluent-Regulated Interfacial Reactivity of Locally Concentrated Ionic Liquid Electrolytes on Lithium Metal
Locally concentrated ionic liquid electrolytes (LCILEs), formed by introducing non-coordinating diluents into ionic liquid systems, …
Spontaneous penetration of carbon nanotubes through lipid bilayers: A computational perspective
Carbon nanotubes (CNTs) have been widely explored for applications ranging from drug delivery to energy conversion. To evaluate their …

Decoding diluent-driven solvation dynamics in locally concentrated ionic liquid electrolytes
The interplay between lithium salts and anions critically influences the electrochemical and physicochemical properties of locally …
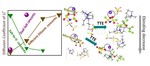
Interfacial impacts of diluent-mediated anion conformational changes in locally concentrated ionic liquid electrolytes
Dilution methods employing weaker-solvating solvents as diluents have shown promise in reducing the viscosity of liquid electrolytes …
Nucleation-controlled doping of II–VI semiconductor nanocrystals mediated by magic-sized clusters
Doping quantum-confined semiconductor nanocrystals offers an effective way to tailor their unique properties. However, the inherent …
Computational insights and phase transition of ruthenium alloy by classical molecular dynamics simulations
Understanding the mechanism of metal solidification holds both theoretical significance and practical importance. In this study, we …

Enhancing electrochemical behavior of localized high-concentration electrolytes solvation structures through antisolvent concentration modulation
Localized high-concentration electrolytes (LHCEs) offer a promising avenue for enhancing the versatility of electrolytes owing to their …
Computational Insights and Phase Transition of Ruthenium Alloy by Classical Molecular Dynamics
Understanding the mechanism of metal solidification is of great theoretical significance and has practical importance. In this study, …
Computational insights and phase transitions of ruthenium alloy using classical molecular dynamics
Evolution in melt structures with temperature during deep undercooling, forming uniform melt-free crystal sites, and the effect of the …